Chronic Myeloid leukemia (CML) belongs to the group of myeloproliferative neoplasms. It is a malignancy of the hematopoietic stem cells with excessive proliferation of the myeloid lineage (especially granulocytes). It is caused by a cytogenetic aberration (Philadelphia chromosome 22) that results in the formation of a BCR-ABL fusion gene. The increased activity of this gene’s product – a tyrosine kinase promotes unregulated proliferation of myeloid progenitor cells, which eventually differentiate into mature cells.
Epidemiology
- Accounts for 15-20 % of adult leukemias.
- 70% of all Leukemias in India are CML.
- Age : The CML is Leukemia of middle age 50-60 yrs.
- Can also be seen in children/adolescents.
- Gender : Slightly higher incidence in males
- M:F ratio – 1.3:1
Symptoms
Asymptomatic
- The onset of CML in insidious
- It may take months to years to manifest completely
Symptomatic
- SOB – Due to anaemia
- Abdominal discomfort – Due to massive splenomegaly
- Lymphadenopathy – Only at times of blast crisis
- Weight loss
- Fever and excessive sweating during sleep (night sweats).
- Headache (uncommon) – Due to hyperleukocytosis
- Bruising and bleeding (uncommon)
- Priapism – Due to leukocytosis
- Pallor
- Retinal hemorrhage
- Gout – There is a high cell turnover rate giving a high urate
- Dyspnea and cough – Due to pulmonary leukostasis, i.e. too many white cells in the blood causing hyperviscosity
Massive Splenomegaly
- Enlarged spleen (2630 gm: normal : 150 to 200 gm ) with greatly expanded red pulp stemming from neoplastic hematopoiesis.
- Extramedullary Hematopoiesis can also produce mild hepatomegaly and lymphadenopathy.
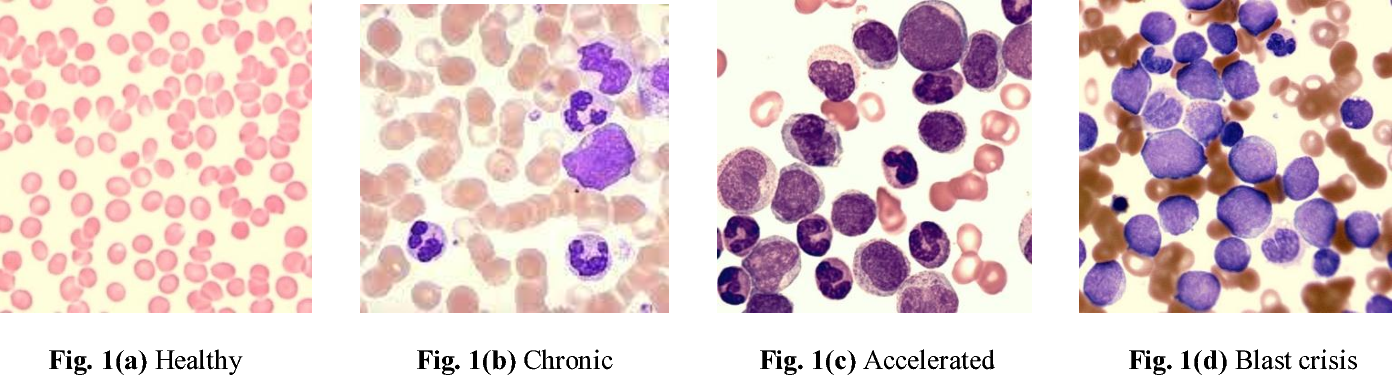
Clinical Phases
CML has three distinct clinical phases –
Chronic Phase
- Most patients are diagnosed in this phase.
- Fairly mild Symptoms (if any)
- Mature cell proliferation.
- Less than 10% blasts in bone marrow.
- M:E ratio is 20: 1
- Blasts 2-5%; basophilia, eosinophilia, monocytosis, thrombocytosis, anemia.
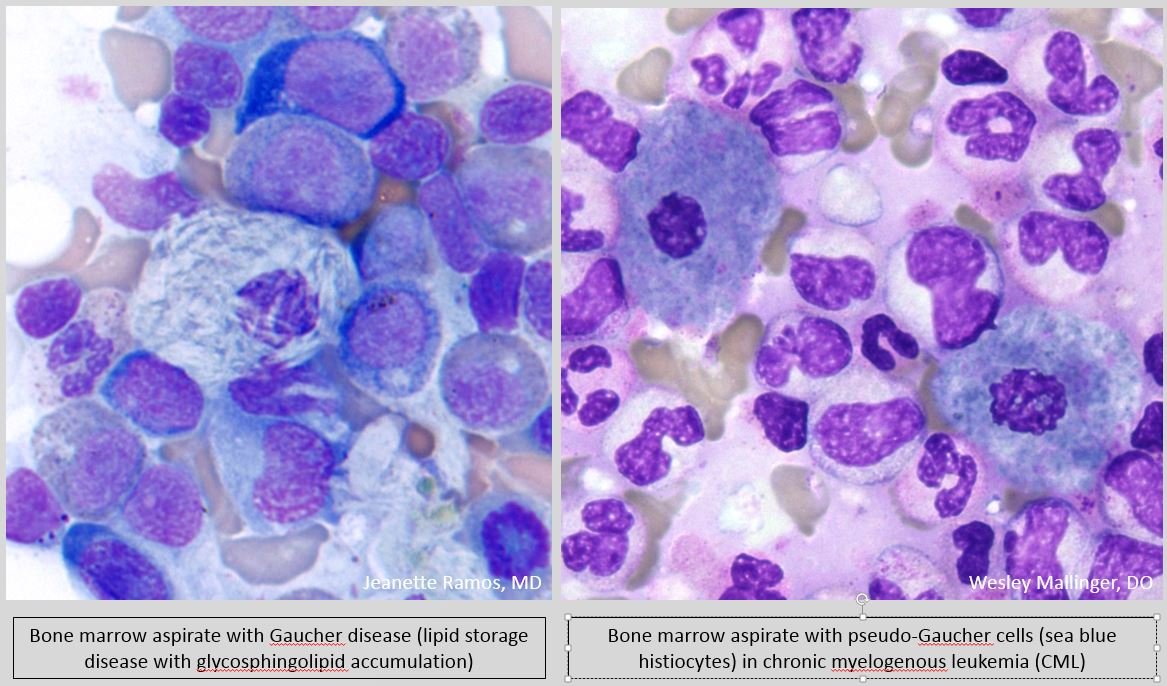
- Large histiocytes (Pseudo-Gaucher cells) with blue granules (Sea blue histiocytes).
- Decreased neutrophil alkaline phosphatase ( NAP Score).
Accelerated Phase
- Patients are considered to be in accelerated phase if any of the following are true –
- 10-19% but less than 30 % blasts
- Basophils >20% of blood.
- Blasts / promyelocytes combined make up 30% or more of blood.
- Thrombocytopenia
- Fever, night sweats, weight loss
Blast Crisis
- Within 6 – 12 months, accelerated phase terminates in blast crisis
- Lymphadenopathy, chloromas
- Peripheral and marrow blasts >20%
- Hyposegmented neutrophils appear(Pelger-Huet anomaly)
- Increased neutrophil alkaline phosphatase ( NAP Score )
Pathophysiology
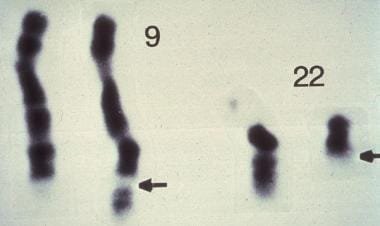
Philadelphia chromosome
- Reciprocal translocation between chromosome 9 and chromosome 22 → formation of the Philadelphia chromosome t(9;22) → fusion of the ABL1 gene (chromosome 9) with the BCR gene (chromosome 22) → formation of the BCR-ABL gene → encodes a BCR-ABL non-receptor tyrosine kinase with increased enzyme activity
- Result: inhibits physiologic apoptosis and increases mitotic rate → uncontrolled proliferation of functional granulocytes.
Genetic changes and clinical course
- Additional chromosomal changes and mutations of tumor suppressor genes and oncogenes (p53, Rb1, or Ras), which emerge during the course of the disease, are responsible for the progression from chronic to accelerated phase and, ultimately, the transition to acute leukemia.
Investigations
The diagnosis of CML is made on the basis of clinical features along with peripheral blood & bone marrow findings and chromosomal study.
Peripheral Blood Findings
- Anemia
- Marked Leukocytosis : Invariably it is more than 100000/ul sometimes may reach up to 500000/ul of blood.
- Platelets : In the initial phase the platelets count may be high but in later stages it becomes subnormal : Thrombocytopenia.
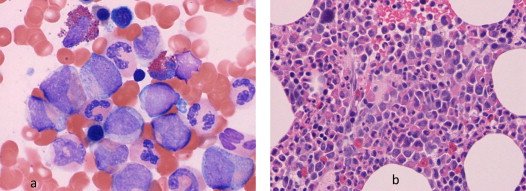
Bone Marrow Aspirate Findings
- Is Hypercellular.
- Erythroid precursors – Normal or Decreased.
- ME Ratio – Very high may be even 50 : 1.
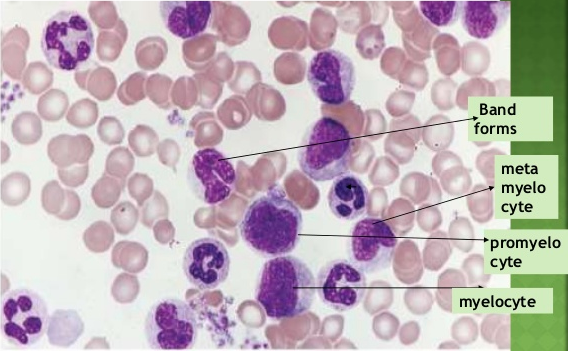
- Proliferation of middle order cells – Myelocytes & Metamyelocytes.
- Characteristic finding is the presence of scattered macrophages with abundant wrinkled, green-blue cytoplasm so called “Sea-blue histiocytes“.
- Increased deposition of reticulin is typical, but overt marrow fibrosis is rare early in the course.
Peripheral Smear Findings
- Most of the time only the peripheral smear is sufficient for the diagnosis of CML.
- RBC – Appear normocytic, normochromic.
- WBC –
- Granulocytes count is very high mainly mature neutrophil ( Characteristic / Predominant cells are “Myelocytes/Metamyelocytes” ).
- Myeloblasts are few.
- Lymphocytes reduced in number.
- Monocyte count may be high.
- Basophilia in CML
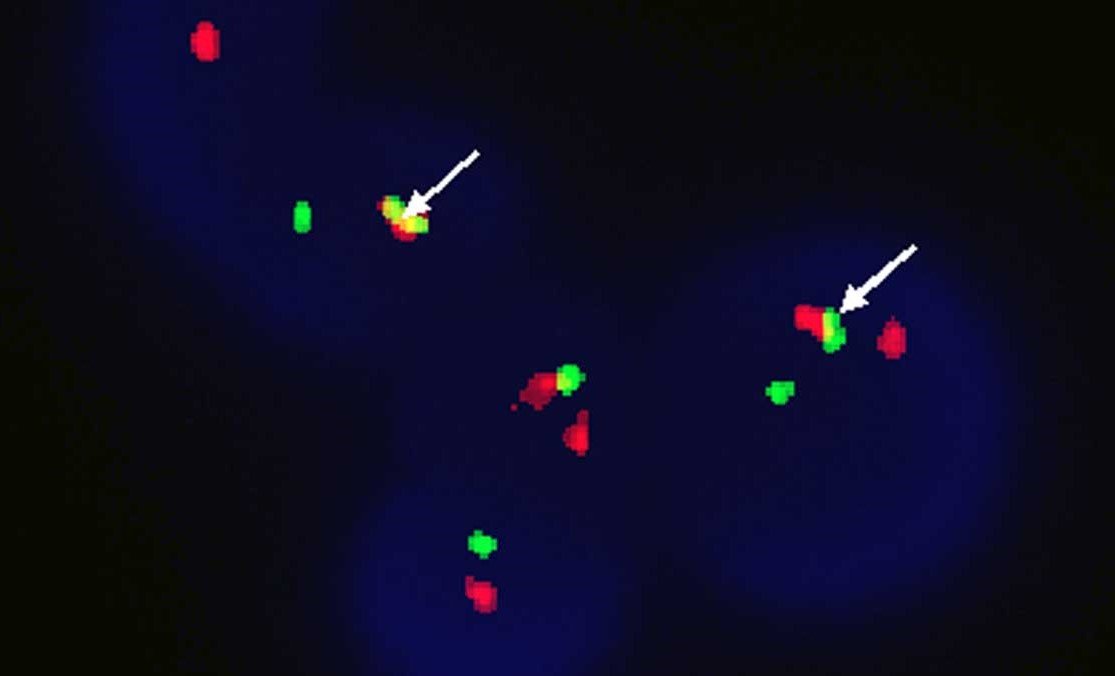
Chromosomal Study
FISH – Used to look for the cytogenetic abnormality; cytogenetics will show Philadelphia chromosome in 97% of cases
Treatment
The goal of chronic myelogenous leukemia treatment is to eliminate the blood cells that contain the abnormal BCR-ABL gene that causes the overabundance of diseased blood cells.
Targeted drug therapy
- Dasatinib (Sprycel)
- Nilotinib (Tasigna)
- Bosutinib (Bosulif)
- Ponatinib (Iclusig)
Side effects of these targeted drugs include swelling or puffiness of the skin, nausea, muscle cramps, fatigue, diarrhea and skin rashes.
Blood tests to detect the presence of the BCR-ABL gene are used to monitor the effectiveness of targeted drug therapy. If the disease doesn’t respond or becomes resistant to targeted therapy such as omacetaxine (Synribo), or other treatments.
Bone marrow transplant
- old age of patient
- disease in acute phase
- poor HLA compatibility of donor